A Imunoinformática Estrutural
Conforme apresentado na primeira parte deste post, em 2009 o NBLI teve
um projeto aprovado no Grand Challenges
Explorations (GCE) da fundação Bill & Melinda Gates. O grupo recebeu um
auxílio de U$ 100.000 dólares para desenvolver um banco de dados de estruturas
peptídeo:MHC (pMHC-I), voltado à identificação de padrões estruturais nestes
complexos. No ano seguinte, o NBLI publicou um trabalho descrevendo padrões
estruturais apresentados pelos epitopos quando complexados à fenda do MHC (Antunes e cols., 2010),
padrão este que não era dependente da sequência do epitopo e que era específico
para cada alotipo1 de
MHC (Figura 1).
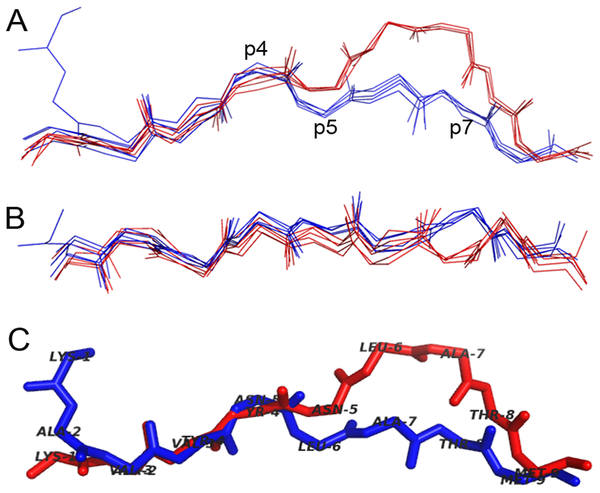
Figura
1. Exemplo da sobreposição de epitopos cristalografados na fenda de MHCs murinos. As estruturas de cinco epitopos
restritos a H2-Db (1CE6, 1S7V, 1WBZ, 1ZHB e 3BUY) e cinco epitopos
restritos a H2-Kb (1FO0, 1FZJ, 1LK2, 1RJY e 1S7R) foram sobrepostas.
(A) Visão lateral salientando as diferenças conformacionais apresentadas por
epitopos no contexto do H2-Db (vermelho) e do H2-Kb (azul),
especialmente entre as posições 5 e 7 (p5-7). (B) Visão superior. (C) Sobreposição
de duas estruturas cristalográficas (1S7V e 1S7R) apresentando o mesmo epitopo (KAVYNLATM)
no contexto destes dois alotipos distintos. Modificado de Antunes e cols., 2010.
Este trabalho foi realizado a partir da análise de
todas as estruturas de pMHCs até então determinadas experimentalmente para alguns
dos alotipos de MHC de classe I mais estudados (H2-Db, H2-Kb,
HLA-A*02:01 e HLA-B*27:05), disponíveis no ProteinData Bank. Além do caráter descritivo, a identificação destes padrões
permitiu o desenvolvimento de uma nova estratégia de predição de estruturas de pMHC-I. Trabalhos recentes têm utilizado ferramentas de ancoramento molecular (docking) para a predição estrutural de
complexos pMHC (Bordner, 2006; Antunes, 2010; Khan, 2010). O docking é amplamente utilizado para
predizer o modo de ligação de fármacos e pequenos ligantes ao sítio ativo de
enzimas e receptores proteicos, apresentando resultados rápidos e confiáveis (Morris, 2009;
Trott, 2010). No entanto, sua acurácia decai rapidamente com o aumento do
número de ligações flexíveis do ligante, o que aumenta o número de conformações
possíveis e eleva o custo computacional para a resolução do problema (Chang e cols., 2010). Enquanto a maioria dos algoritmos disponíveis apresenta resultados confiáveis
para o ancoramento molecular de ligantes com até 10 ligações flexíveis, um
peptídeo típico (apresentado por MHC de classe I) possui cerca de 9 aminoácidos
e pode apresentar mais de 40 ligações flexíveis. Neste contexto, a descoberta
de padrões conformacionais específicos para cada alotipo de MHC viabiliza o docking de complexos pMHC-I, na medida em
que permite reduzir o número de ligações flexíveis do ligante. Esta foi a
premissa utilizada no desenvolvimento de uma nova estratégia de predição
estrutural de complexos pMHC-I, referida como D1-EM-D2
(Figura 2).

Figura 2. Fluxograma da
abordagem D1-EM-D2. A estrutura de um dado complexo pMHC-I, cuja estrutura ainda
não é conhecida, pode ser modelada em três etapas: (i) Docking, (ii) Energy
Minimization e (iii) Docking (D1-EM-D2).
A sequência linear de aminoácidos de um peptídeo pode ser utilizada como
entrada para um script do PyMOL (a), o qual gera um arquivo de coordenadas
espaciais (PDB) para o epitopo alvo, ajustando a conformação da cadeia
principal ao padrão “alotipo-específico”. Uma estrutura de referência contendo
o alotipo de interesse é utilizada como “Doador de MHC”, removendo-se o
peptídeo da fenda. O “Doador de MHC” e o “Epitopo 3D” são utilizados como
entrada para o ancoramento molecular (docking) com o Autodock Vina (b), gerando
um novo pMHC-I. Um ciclo de otimização da estrutura é realizado para ajustar o “Doador
de MHC” ao novo epitopo. Modificado de Sinigaglia e cols., 2013.
Mesmo considerando-se apenas os genes
clássicos de MHC em humanos (HLA-A, HLA-B e HLA-C)2,
já são descritos mais de 8.000 alelos3,
dos quais uma parcela insignificante possui estrutura conhecida. Cada um destes
alelos codifica a cadeia pesada de um alotipo de MHC capaz de apresentar milhares
de peptídeos, muitos dos quais seriam de grande interesse para estudos de
imunologia (variantes virais, autoantígenos, epitopos que apresentam
reatividade-cruzada, etc). Muitos avanços foram feitos no campo da Cristalografia
de Raios-X e da Ressonância Magnética Nuclear, permitindo a determinação experimental
de um número cada vez maior de estruturas proteicas. No entanto, estes métodos
continuam sendo extremamente caros e demorados. Assim sendo, considerando-se a
magnitude e a variabilidade deste sistema, análises estruturais em larga escala de complexos pMHC-I seriam
praticamente inviáveis sem o auxílio de métodos in silico, rápidos, precisos e de baixo custo.
A abordagem D1-EM-D2 foi
amplamente validada em comparação com estruturas cristalográficas, sendo então aplicada na
predição em larga escala de complexos pMHC-I apresentando peptídeos imunogênicos.
Estes complexos foram posteriormente disponibilizados em um banco de dados
específico, o CrossTope (Sinigaglia, 2013). Juntamente com o arquivo contendo
as coordenadas espaciais do complexo, o CrossTope disponibiliza informações
sobre o alelo de MHC, a proteína e o organismo de origem do epitopo
apresentado, etc. São fornecidos links
para bancos com informações complementares, como o IEDB e o NCBI. Além
disso, o banco fornece uma imagem do potencial eletrostático sobre a superfície
do pMHC que fará contato com o TCR. Estudos recentes indicam que características
estruturais desta superfície, como cargas e topografia, podem direcionar o
reconhecimento pelos linfócitos T CD8+ (Sandalova, 2005; Antunes, 2011; Shen, 2013). De acordo com resultados in vitro,
mesmo a troca de um único aminoácido pode abortar o reconhecimento por uma dada
população de linfócitos (Fytili, 2008).

Figura 3. Potencial
eletrostático na superfície de interação com o TCR. A imagem mostra a vista
superior de três complexos pMHC formados por variantes de um mesmo epitopo (com
trocas por alanina), no contexto do HLA-A*02:01. Observe a posição dos domínios
α1 e α2 do MHC, entre os quais se
localiza o epitopo. Regiões com cargas positivas (azuis) e negativas
(vermelhas) são representadas em uma escala de -5 kT a +5 kT. As
superfícies de complexos que estimulam altos níveis de IFN-gamma frente a uma
população de linfócitos específica contra o tipo selvagem (CVNGVCWTV) são
muito semelhantes (A e B), enquanto diferenças de topografia e cargas (setas verdes)
são visíveis na superfície de um complexo que estimula fracamente a mesma
população de linfócitos (C). Modificado de Antunes e cols., 2010.
Além de continuar trabalhando no aperfeiçoamento e automatização da abordagem
para predição estrutural de complexos pMHC-I, nosso grupo tem especial interesse
na predição de reatividade-cruzada entre epitopos virais. Na terceira e última
parte deste post, serão apresentados
resultados referentes ao uso de estatísticas multivariadas para a triagem
virtual de complexos pMHC-I. Esta abordagem baseada em estrutura nos permitiu
predizer uma reatividade-cruzada até então desconhecida, entre epitopos que não
compartilhavam nenhum aminoácido em sua sequência, um resultado que já está
sendo confirmado por experimentos in
vitro.
Post de Dinler Amaral Antunes
NBLI - Núcleo de Bioinformática do Laboratório de
Imunogenética.
Aluno de Doutorado do Programa de Pós-Graduação em Genética
e Biologia Molecular da Universidade Federal do Rio Grande do Sul
(PPGBM/UFRGS).
1 A expressão “alelos
de MHC” é usualmente utilizada para referenciar tanto as variantes dos genes
quanto as diferentes proteínas codificadas por estas variantes gênicas. No
entanto, este segundo uso da expressão não é adequado uma vez que o termo
“alelo” se refere especificamente a DNA, não a proteína. A expressão “alotipo”
é indicada para referenciar as diferentes proteínas codificadas por alelos de
MHC.
2 A cadeia pesada do complexo responsável pela apresentação de peptídeos endógenos na superfície celular é referida genericamente como MHC de classe I (na sigla em inglês para Major Histocompatibility Complex), mas a nomenclatura varia de acordo com a espécie. Por exemplo, em humanos ele recebe o nome HLA (na sigla em inglês para Human Leukocyte Antigen), em camundongos é referido como antígeno H2, etc.
3 Até as 14 horas de Quarta-Feira, dia 07/05/2014, havia 8.124 alelos de MHC de classe I listados no http://hla.alleles.org/nomenclature/stats.html.
Nenhum comentário:
Postar um comentário