Research
Development of computational methods for structural modeling of pMHC complexes.
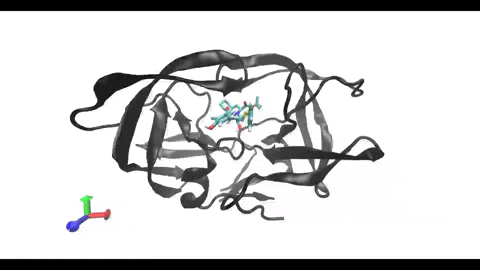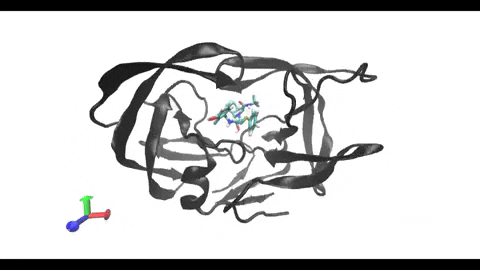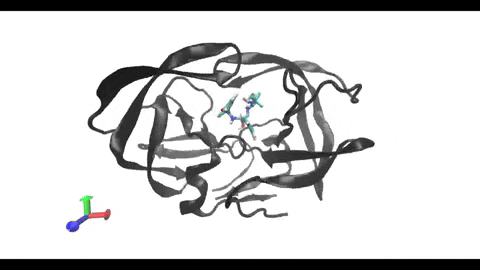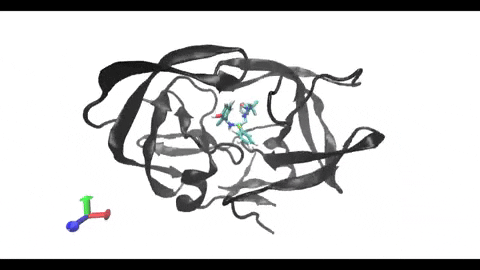
 are present in almost every cell, being involved in the surface presentation of peptides derived from intracellular proteins. This presentation drives the activation of cytotoxic T-cells, triggering a cellular response. On the other hand, class II MHCs are present only in “professional” antigen-presenting cells (phagocytes), being involved in the surface
presentation of peptides derived from extracellular proteins. This presentation drives the activation of helper T-cells, triggering a humoral response. Understanding the structural differences between these two types of MHC receptors and how they bind their ligands is key for the development of better modeling and binding prediction methods. CD, cluster of differentiation. TCR, T-cell receptor. Modified from Antunes et. al, 2019.")
Understanding the mechanisms involved in the activation of an immune response is essential to many fields in human health, including vaccine development and personalized cancer immunotherapy. A central step in the activation of the adaptive immune response is the recognition, by T-cell lymphocytes, of peptides displayed by a special type of receptor known as Major Histocompatibility Complex (MHC). Considering the key role of MHC receptors in T-cell activation, the computational prediction of peptide binding to MHC has been an important goal for many immunological applications. This problem, however, is much harder than most docking problems in drug discovery, given the length and flexibility of the peptide-targets. In order to overcome the high dimensionality of this sampling problem, three strategies have been devised: (i) constrained backbone prediction, (ii) constrained termini prediction, and (iii) incremental prediction. Each of these strategies has advantages and limitations, and over the years I have made contributions in all three categories (see Antunes et. al, 2019). First, I have identified allele-specific patterns that were used to develop a constrained backbone prediction tool called DockTope. DockTope was validated for 4 MHC alleles through cross-docking of available crystal structures, being recently integrated to the IEDB Analysis Resource collection as the first open-acess docking-based webserver for modeling pMHC complexes. Later, I started working with DINC, a meta-docking incremental approach that is suited for docking large ligands. I provided proof-of-concept of its use for general structural prediction of pMHC complexes (i.e., modeling different MHC alleles and peptide lengths). Finally, we implemented a constrained termini prediction method for fast generation of ensembles of bound conformations of pMHC complexes APE-Gen. This method allows for large-scale structural analysis of pMHC complexes, being also applicable to to virtually all known HLAs.
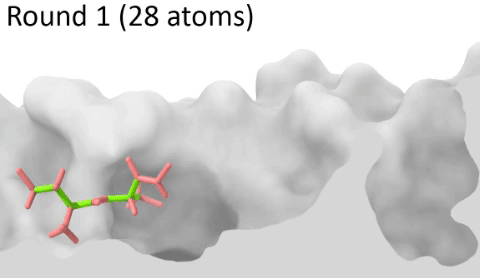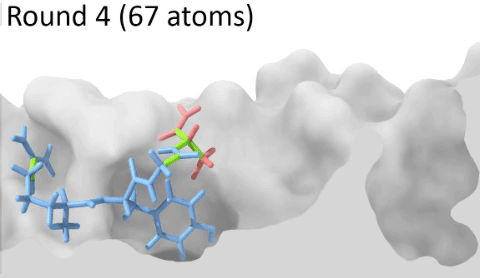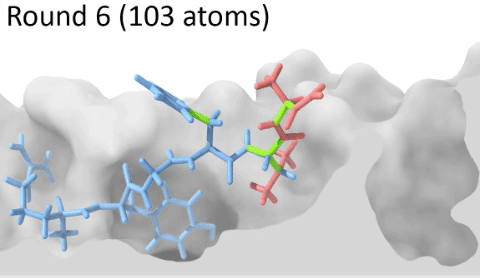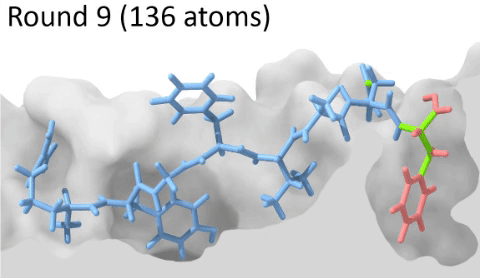
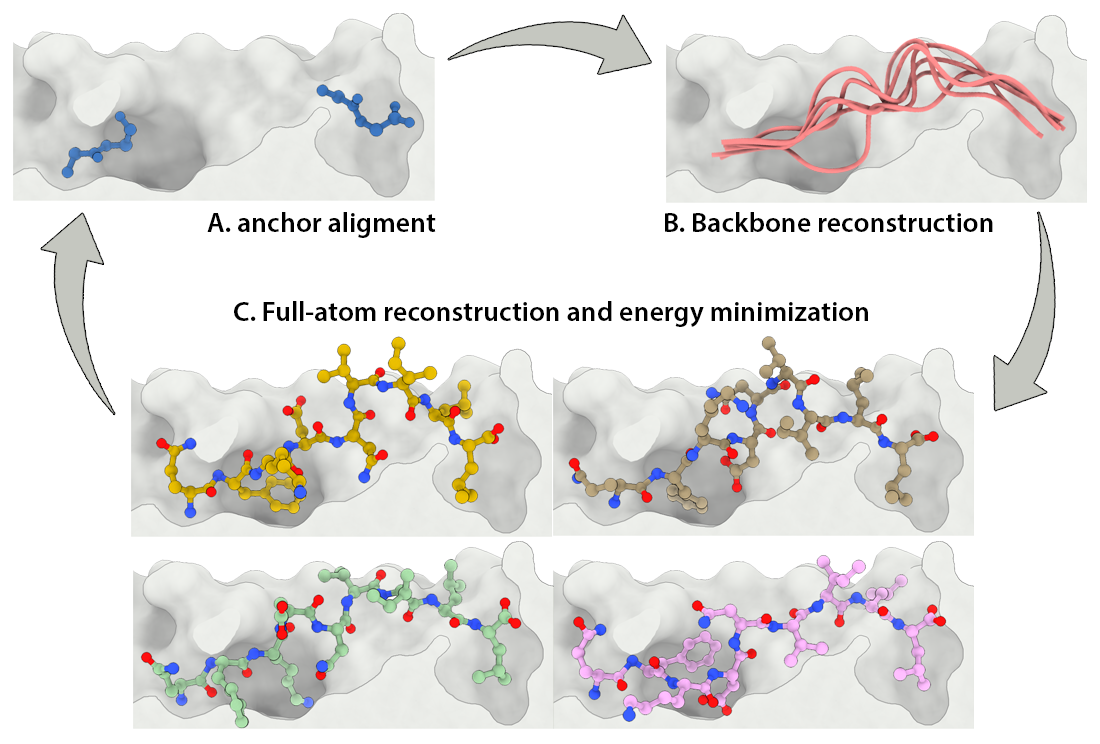
Development of a customizable environment for the structural analysis of peptide-HLA complexes.
Using Jupyter Notebook and Docker, we have created a customizable environment, called HLA-Arena, that enables researchers to easily model any class I pHLA complex of interest and perform varied structural analyses. HLA-Arena includes different workflows, defined as separate notebooks, that consist of the following main stages:
Input processing: Available structures of HLA receptors are obtained from the PDB to be used as such or as templates. Unavailable HLA structures are modeled with Modeller, using a HLA sequence and the structure of a similar HLA receptor as template, if these are provided by the user. Alternatively, users can just provide an allele name (e.g., HLA-A*24:02); HLA-Arena will then fetch the proper sequence from IMGT/HLA, and a reasonable template (based on the HLA supertype classification) from the PDB. In addition, binding affinity of peptides can be estimated with MHCflurry 12 to select the most relevant ones.
Peptide docking: Structures of pHLA complexes are modeled with APE-Gen and/or DINC, which only requires the sequence of the target peptide(s) and the HLA structure(s) obtained previously. Modeled structures can also be minimized with a force field, using OpenMM.
Data analysis: A variety of post-processing options for data analysis can be incorporated in a workflow. These include binding mode rescoring or peptide ranking with DINC, and structure visualization with NGL Viewer, among others.
For a smooth user experience, all computational tools involved in HLA-Arena are packaged within a Docker image, therefore eliminating the burden of managing software dependencies. Another advantage of Docker containerization is to make HLA-Arena platform-agnostic. As a result, it can be deployed on a desktop computer or a high-performance computing cluster, across different operating systems. Users can customize available workflows by adding modeling or analysis steps. We plan to continuously expand the capabilities of HLA-Arena by providing support for additional tools.
 for various
peptides, and these binding modes are scored to rank the peptides with Smina. In the virtual screening workflow, after filtering peptides with MHCflurry, ensembles of binding modes are generated with APE-Gen for the selected peptides, and the top-scoring binding modes are used to rank these peptides with Smina, in terms of binding affinity to a (set of) HLA receptor(s). Note that these workflows can be modified, and that new ones can be created by users. In each application, different types of data analysis can be used to guide the selection of the best pHLAs before experimental validation. Modified from [Antunes *et. al*, 2020](https://dinlerantunes.com/publications/antunes2020-hla-arena.pdf).")
Research during the COVID-19 pandemic
DINC-COVID webserver for ensemble docking
, formed by the combination of both protomers. **1**. Ensemble generation: Three different ensembles of the dimeric receptor are pre-computed (e.g., from available crystal structures or molecular dynamics simulations) and made available through the DINC-COVID webserver. Different shades represent different receptor conformations within each ensemble. **2**. Input selection: The user can select one of the available ensembles, and upload the ligand of interest (e.g., drug or peptide). **3**. Ensemble docking: The parallelized meta-docking approach DINC is used to sample alternative binding modes using each of the receptor’s conformations included in the ensemble. **4**. Scoring and Ranking: All generated binding modes are rescored and ranked according to three different scoring functions (e.g., Vina, Vinardo and AutoDock4). **5**. Output complexes: A number of top scoring complexes are returned to the user, reflecting the flexibility of both the ligand and the receptor.")
The novel coronavirus SARS-CoV-2, which causes the respiratory disease COVID-19, went from an outbreak to a world-wide pandemic in just a few months. In response, there have been unprecedented global efforts to develop effective treatments. Among pharmacological targets, proteins involved in the viral replication have been used in several computational studies focused on drug design, drug repurposing and virtual screening. Unfortunately, proposed SARS-CoV-2 inhibitors have not yet impacted the course of the COVID-19 pandemic. Most of these efforts, however, have largely ignored the issue of receptor flexibility. In this context, we have implemented a computational tool for ensemble docking with SARS-CoV-2 proteins, including the main protease (Mpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp). DINC-COVID is available as a user friendly webserver, providing plausible binding modes between conformations of a selected ensemble and a user uploaded ligand. These binding modes are sampled with DINC, our parallelized meta-docking tool, and scored with three different scoring functions.
Molecular dynamics of Compstatin analogs
On another study, I used molecular dynamics simulations to uncover the mechanistic properties of several compstatin analogs. Compstatin is a peptide-based drug proven to be a very promising inhibitor of the complement system, an important component of the innate immunity. A recent compstatin analog is being developed as a candidate drug against several pathological conditions, including COVID-19. However, the reasons behind its higher potency and increased binding affinity to complement proteins are not fully clear. I performed simulations involving six analogs alone in solution and two complexes with compstatin bound to complement component 3. These simulations reveal that all the analogs we consider, except the original compstatin, naturally adopt a pre-bound conformation in solution. Interestingly, this set of analogs adopting a pre-bound conformation includes analogs that were not known to benefit from this behavior. We also show that the most recent compstatin analog forms a stronger hydrogen bond network with its complement receptor than an earlier analog.
 is elongated and u-shaped. The conformation of 4W9A (bound to C3c, under PDB code 2QKI) is more compact and α-shaped. The conformation of Cp10 (free in solution, extracted from an NMR ensemble) is more compact than that of 1A1P. The disulfide bridge creating the cycle is represented with thin lines in all analogs.")
Identification of broadly-protective SARS-derived peptide-targets
Fortunately, we have now multiple effective vaccines that are already been used to control the ongoing COVID-19 pandemic. Most of these vaccines aim at inducing the production of neutralizing antibodies against envelope proteins. However, envelope proteins are knowingly more susceptible to selection pressure, and therefore more prone to mutations that can quickly lead to resistance to treatment. In other words, even if successful vaccination campaigns are executed in different countries, it is unclear for how long this immunization will be effective. In addition, the large reservoir of SARS-type viruses in the wild highlights the continued risk for new pandemics in the future. Therefore, there is a need for effective vaccination strategies that would protect individuals against a broader range of SARS-like coronaviruses. To address this problem, we have started working on the creation of a new HLA-Arena workflow specifically designed to help the identification of conserved targets across SARS-CoV-like viruses. This project was funded through the IIBR:Informatics:RAPID award mechanism of NSF.
 protein for SARS-CoV-2, SARS-CoV-1 and other SARS-like coronaviruses will be aligned. A sliding window procedure will be conducted to evaluate the conservancy of every overlapping peptide in the region of interest. The most conserved regions will be selected for a consensus HLA-binding analysis, using both sequence-based and structure-based methods. Finally, a surface analysis will be conducted to reveal the potential impact of mutations over T-cell recognition.")
Identification of structural features driving T-cell cross-reactivity.
 using structural data from modeled peptide-MHC complexes. In agreement with experimentally-observed patterns of cross-reactivity among HCV genotypes, complexes presenting peptides from HCV genotypes 4, 5 and 6 were grouped with the optimal responders from genotype 1. Complex G3-18, the only complex with no measurable T-cell response, was placed in a completely separated position in the scatter plot. Other complexes from genotypes 2 and 3 (weak response), are also separate from strong responders. Modified from Antunes et al., 2011.")
Using DockTope, I have investigated the structural similarity of pMHC complexes presenting cross-reactive and non-cross-reactive variants of the immunodominant peptide NS3-1073, derived from Hepatitis C Virus (HCV). This HLA-A*02:01-restricted peptide was included in a vaccine that was protective only against certain HCV genotypes (cross-genotype-reactivity), according to a study previously performed by a German group (Fytili et al., 2008). Applying Principal Component Analysis (PCA) and hierarchical clustering on data extracted from modeled pMHC complexes, I was able to show that observed patterns of cross-reactivity were mostly driven by structural similarity between the complexes (see Antunes et. al, 2011); particularly, topography and charge distribution over the T-cell-interacting surface. Using this knowledge, I executed a virtual screening against a panel of unrelated viral-derived targets, also modeled in the context of HLA-A*02:01. This analysis indicated potential cross-reactivity of the wild-type HCV-derived peptide (NS3-1073) with peptides from Epstein-Barr Virus, Influenza and HIV. Some of these peptides had little or even no sequence identity with the wild-type HCV peptide, making these cross-reactivities impossible to be predicted using sequence-based analyses. All these targets were later tested with lymphocytes from HCV-infected patients and healthy vaccinated individuals, confirming the predicted cross-reactivities (see Zhang et. al, 2015). More importantly, cross-reactivities with these heterologous targets were associated to differential response to vaccination, highlighting the importance of this issue for vaccine design. More recently, I extended these analyses to evaluate previously described “cross-reactivity networks” among virus-derived peptides. I used structure-based clustering of modeled pMHC complexes to help explain apparent inconsistencies in reported cross-reactivities, and proposed testable hypotheses on the implications of pMHC structural similarity to T-cell cross-reactivity and cancer immunotherapy (see Antunes et. al., 2017).
. Colors indicate the range of the electrostatic potential over the surface, from −5 kT/e (red) to +5 kT/e (blue). Complex information and peptide sequence are depicted below each pMHC. For crystal structures, the corresponding PDB ID is also provided. Complexes with no published crystal structure were modeled with DockTope. Peptide sequences indicate mutations in relation to the reference peptide (central column). The green arrow highlights an \"outstanding\" features of the peptide with \"limited\" cross-reactivity (left column). Black and gray arrows indicate the intensity and preferred directionality of cross-reactivities observed in vitro. Modified from Antunes et. al, 2017.")
CrossTope: A structural database for cross-reactivity assessment

The Structural Data Bank for Cross-Reactivity is a curate repository of three-dimensional structures of pMHC complexes, focused on immunogenicity, similarity relationships and cross-reactivity prediction. We used DockTope to predict more than 500 unknown pMHC structures, now publicly available through the CrossTope Data Bank.
A new classification method to understand available docking strategies accounting for protein flexibility.
 and ligands (small objects) illustrate the level of flexibility considered in each docking category. Modified from Antunes *et. al*, 2015.")
Molecular Docking became an essential tool for research in drug design, and several different software are currently available. Older applications explored only flexibility of the ligand, while keeping the protein rigid through the entire search. In many cases this approach would not be enough to reproduce the correct protein-ligand binding mode, since proteins are extremely flexible and can change the conformation of the binding site. Nowadays, most docking methods would consider some level of protein flexibility during the search and most classification attempts would relate these different methods to one of the main biomolecular recognition models (induced fit or conformational selection). However, there exists a great diversity of docking methods accounting for protein flexibility, and any classification based on a dichotomy between these two theoretical models is bound to fail. Contrary to what is frequently done, I proposed a more algorithmic classification, focusing on the level of protein flexibility accounted for (e.g. implicit or explicit, partial or full). This alternative classification should help new users to navigate through all the diversity of docking approaches, allowing them to choose the one the best suits the research problem they want to investigate (see Antunes et. al, 2015).
Identification of structural features involved in resistance to HIV-1 protease-inhibitors.
 of this simulation indicates three \"islands\" of low energy conformations (i1, i2 and i3), from which different structures were recovered (NF-i1, NF-i2, NF-i3). Altered conformations of the ligand impact its ability to bind the target enzyme. Modified from Antunes et. al, 2014.")
The Human Immunodeficiency Virus type 1 protease enzyme (HIV-1 PR) is one of the most important targets of antiretroviral therapy used in the treatment of AIDS patients. The success of protease-inhibitors (PIs), however, is often limited by the emergence of protease mutations that can confer resistance to a specific drug, or even to multiple PIs. Using molecular docking and molecular dynamics, I evaluated the impact of two unusual mutations (D30V and V32E) over the dynamics of the PR-Nelfinavir complex (see Antunes et. al, 2014). These mutations were identified in drug free HIV-1 patients (from Porto Alegre, Brazil), and involved codons that were previously related to major drug resistance to Nelfinavir. Both studied mutations presented structural features that indicate resistance to Nelfinavir, each one with a different impact over the interaction with the drug. The D30V mutation triggered a subtle change in the PR structure, which was also observed for the well-known Nelfinavir resistance mutation D30N, while the V32E exchange presented a much more dramatic impact over the PR flap dynamics. Moreover, this in silico approach was also able to describe different binding modes of the drug when bound to different proteases, identifying specific features of HIV-1 subtype B and subtype C proteases. A better understanding of the differences among HIV-1 subtypes and the molecular features involved in drug-resistance will allow physicians to prescribe the most effective drug for each individual patient, avoiding treatment failure and promoting durable remission of HIV-1.